2024. 10. 26. 15:03ㆍLab skills
Animal cell culture can be broadly divided into animal cells, culture media, culture methods, and storage methods.
Animal cells
Animal cells are divided into primary cells with a limited lifespan isolated from animal tissue or blood, and (cancer) cell lines that can proliferate indefinitely.
“Primary cells” are cells that have completed differentiation and do not proliferate, or can proliferate only for a certain period of time, and then die through the aging process. Less differentiated cells, such as epithelial cells or vascular endothelial cells of tissues that are easily damaged and repaired, such as skin or gastrointestinal tract, fetal cells, or stem cells, are relatively easy to proliferate, and are therefore used in various ways to study the functions of normal cells.
“(Cancer) cell lines” refer to cells that are directly isolated from cancer tissue and proliferated from a single cell, or primary cells that have been immortalized (cancerized) to enable indefinite proliferation. Cancer cell lines can be continuously used by subculture, and since the same characteristics are maintained, the reproducibility of experimental results is high, so they are used for research on the characteristics of cancer cells or for functional studies after introducing specific genes. Cancer cell lines are distributed from cell line banks, and the passage number of the cell line is recorded during passage culture.
The first question is, why is it necessary to record the “passage number” of cancer cells that can proliferate infinitely?
Before explaining the reason, let’s learn about the origin of “HeLa cells,” one of the most commonly used cancer cell lines. HeLa cells are cancer cells isolated from a cervical cancer patient named Henrietta Lacks by George Otto Gey in 1951, and the cell line name is derived from the patient’s name. The HeLa cells we use in 2020 were created 70 years ago and spread to countries around the world, and have been used under the same name while passage cultured to this day.
So are these HeLa cells the same cells that were isolated in 1951? It is easy to predict that they are genetically quite different cells, except for the name. In order for cells to proliferate, DNA replication must occur. Although cells have sophisticated DNA replication systems and mutation prevention systems, numerous mutations must have accumulated over time as they proliferated. That is, the HeLa cells from 1951 and the HeLa cells used in any laboratory on Earth today are genetically absolutely not the same.
According to a 2013 Nature paper, when comparing and analyzing HeLa cells used in different laboratories, distinct chromosome duplications and rearrangements were confirmed, confirming that each cell line was not the same. The error rate of DNA polymerase is about 10-8 to 10-10, and the human genome consists of 6.4x109 nucleotides, so it is calculated that approximately one mutation can occur for each replication. Statistically, one mutation may seem like an insignificant number, but it cannot be ignored when considering the fact that sickle cell anemia is caused by only one mutation [GAG (Glu)->GTG (Val)]. It may
vary depending on the purpose of the experiment, but at least in order to secure the reproducibility of experiments conducted in each laboratory, a strategy to minimize the occurrence of mutations in cell lines while they are used in that laboratory is necessary. The method is to receive a cell line from a cell line bank, use the initial cultured cells as parent cells to create a master cell stock (GMP, master cell bank (MCB)), and use the cells proliferated from the master cell stock to create a working cell stock (GMP, working cell bank (WCB)). After a certain number of passages, cells proliferated from a new master cell stock or another working cell stock are used. In this way, by limiting the passage number after distribution, mutation accumulation can be minimized, thereby ensuring the reproducibility of the experiment.
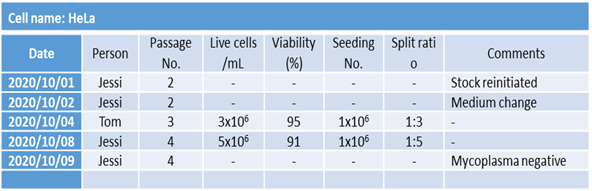
In other words, even if the cell lines used in each laboratory are not identical, it is necessary to record the passage number to monitor the number of times the cell line has been subcultured in order to minimize changes in the characteristics of the cell line due to mutations after distribution and to ensure “experimental reproducibility.”
Suspension cells and adherent cells
Animal cells can be divided into “suspension cells” and “adherent cells” depending on how they grow.
“Suspension cells” are cells that do not need to attach
to a surface to proliferate, and most immune cells in the blood belong to this category. “Adherent cells” are cells that need to attach to a surface to proliferate, and cells derived from most tissues, including epithelial cells and fibroblasts, are adherent cells.
If adherent cells are prevented from attaching, the cells will not proliferate and will die. So what does cell attachment mean? The cell membrane of adherent cells expresses various surface proteins, among which adhesion molecules involved in cell adhesion are involved in the extracellular matrix (ECM) or cell-to-cell binding in vivo.
This binding also plays an important role in cell signaling. Among cell culture vessels, those that are marked as “cell culture treated” mean that the surface has been made suitable for cell culture, and are mainly treated to have a “positive charge.” Since cell surfaces generally have a negative charge, cell attachment is promoted. Since cell attachment is involved in various activities such as cell proliferation, differentiation, and migration, it is important to create an environment suitable for attachment. If representative ECM components such as collagen, fibronectin, laminin, and gelatin are coated on the bottom of the culture vessel, cell growth is promoted. Using a vessel coated with ECM components can minimize the detachment of cells with weak adhesion.
Culture medium
Culture medium contains various nutrients, growth hormones, vitamins, salts, trace elements, etc. necessary for cell proliferation. In addition to chemical nutrients, culture media use fetal bovine serum (FBS), which contains various growth promoting factors, as an additive. In cell culture for protein production or pharmaceutical manufacturing purposes, culture media composed only of chemical substitutes are sometimes used to minimize contamination of the components contained in FBS. Since the cell line bank provides optimized media information for each cell line, you can follow it. However, depending on the purpose of the experiment, a process of optimizing the media is necessary.
When producing recombinant proteins, the proportion of recombinant proteins, which are foreign proteins, among the total proteins synthesized by each cell line is significant, and the composition of amino acids constituting each recombinant protein is also different. Since the protein cannot be produced even if only one of the 20 amino acids constituting the recombinant protein is depleted first, media optimization for each cell line for recombinant proteins is necessary.
Second question: Why is the FBS added to the media “heat inactivated” before use?
FBS contains various components necessary for cell growth, as well as complement, an immune factor that binds to antibodies and causes cytotoxicity, and various inhibitors. Heat inactivation is performed at 56°C for 30 minutes, inactivating complement and inhibitors, thereby removing potential cytotoxicity-inducing and growth-inhibiting factors.
Heat-inactivated FBS should be used for cytotoxicity assays using antibodies or for culturing teratocarcinomas, ES cells, or insect cell lines. However, since some growth factors may also be inactivated by this heat treatment, experiments where the necessity is not mentioned or long-term heat treatment at higher temperatures should be avoided.
Cell culture method
It is important to use a medium suitable for each cell line for animal cell culture, seed an appropriate number of cells, and culture for an appropriate period of time.
Floating cells can be relatively easily recovered through centrifugation and subcultured. On the other hand, adherent cells are attached to the surface of the culture dish through adherens junctions (AJ) and bind to adjacent cells through tight junctions (TJ). The process of detaching adherent cells during subculture is the most important part because it affects cell viability and subsequent experimental results.
Let's take a closer look at the process of detaching adherent cells during subculture.
To detach adherent cells from a culture dish, first, the medium is completely removed, washed with DPBS (Ca2+, Mg2+ free), and then treated with Trypsin/EDTA.
At this time, the DPBS washing step also plays an important role in detaching cells.
Attachment of cells involves attachment proteins (AJ and TJ), and attachment proteins require Ca2+ ions as a cofactor. The medium contains a large amount of Ca2+ ions, which are cofactors of the attachment proteins, and FBS contains trypsin inhibitors called anti-trypsin and alpha-2-macroglobulin. The DPBS washing step removes the Ca2+ ions and trypsin inhibitors remaining in the medium, thereby helping the Trypsin/EDTA action.
EDTA is a chelator that adsorbs metal ions such as Ca2+, and by adsorbing and removing Ca2+ ions from the attachment protein (TJ), it changes the molecular structure and causes the cells to detach.
Trypsin is a serine protease that recognizes and cleaves the C-terminal of Arginine or Lysine (--X-Arg(Lys)-/cleavage site/-X--) among the attachment protein sequences on the surface of cells attached to the culture dish, thereby detaching the cells from the culture dish.
The trypsin treatment time needs to be optimized depending on the type of cell. DPBS, Trypsin/EDTA, and media are used after warming them in a 37°C constant temperature water bath, and the treatment time is optimized for 3 minutes. It is best when the cells are almost round and fall off naturally when lightly tapped. If the trypsin treatment time is longer than necessary, it will excessively damage the cell surface proteins, lowering the cell viability rate and adversely affecting cell surface protein analysis.
After trypsin treatment, the medium containing FBS is added, because the trypsin inhibitor contained in FBS stops the enzymatic action of trypsin.
The third question is, why is it that cell lines are only cultured until they are 70-90% confluent?
When cells grow confluently, “contact inhibition” and cell growth arrest occur. Unlike living organisms that continuously supply nutrients and remove waste products through blood, waste products, mainly lactic acid, are locally accumulated around the cells under in vitro culture conditions. Acidification of the medium reduces the stability of the genome and increases the occurrence of mutations due to DNA damage. Also, after contact inhibition occurs, the cell proliferation rate and overall activity decrease, which affects the results of subsequent studies, so it is important to culture at an appropriate density.
Another question is, when culturing cells, the medium is exchanged once every 2-3 days, but if the cell density is low, can the medium be exchanged every 5-10 days? The answer is “No.”
The half-life of the growth factors contained in FBS among the medium components is not long under culturing conditions at 37℃. The medium exchange cycle is related not only to the acidification of the medium due to cell growth, but also to the activity of the medium components. Therefore, if the number of available cells is small, it is recommended to reduce the size of the culture vessel to maintain the cell density at an appropriate level, and to maintain the medium exchange cycle at 2-3 day intervals.
cell culture
Experiments using cells, it is very important to accurately measure the number of cells and use a certain number of cells to obtain reproducible results. For example, if the density of cells is too high, they compete with each other for the medium and the rate of waste accumulation also increases, but if the number of cells is appropriate, the growth factors secreted by the cells help proliferation, but if the number of cells is too small, it is difficult to expect this synergistic effect. Since cell sensitivity to drugs increases at low densities compared to optimal densities, it is important to maintain an appropriate level of cell density when evaluating the activity of cytotoxic drugs.
Solving microbial contamination problems in animal cell culture
1. Determine whether contamination is a problem with the culture or the cell line and respond accordingly.
- The first principle for solving microbial contamination is to discard the culture.
- If there is a new stock, culture using the new stock.
(The new stock can be the same lot or a different lot. If the same lot and a different lot are cultured at the same time and only the same lot has a problem, discard the entire problematic lot and use the other lot.)
- If there is no problem with the new stock, continue to use it. If contamination is confirmed in the new stock, discard or proceed with the microbial removal step depending on the cell type.
2. In cases where it is difficult to discard, such as a stable cell line, and the entire stock is contaminated, consider removing the microorganisms with antibiotics.
- Identify the characteristics according to the type of microbial contamination.
1) Bacterial contamination
- This can occur if no antibiotics are used in the culture medium.
- There is a possibility of dual-resistant microorganism contamination even when Penicillin/Streptomycin (P/S) is used.
- It may appear dirty under a microscope and become cloudy along with a change in the color of the medium.
- If dual-resistant microorganism contamination is suspected, an antibiotic for microorganisms with a different mechanism, such as Gentamicin, can be added.
2) Mycoplasma contamination
- The growth rate may be slow, the cell shape may change, and it may appear dirty under a microscope.
- Since Mycoplasma grows slowly, it does not show symptoms of a rapid change in the color of the medium.
3) Yeast/Fungi contamination
- If an antibiotic for bacteria such as P/S is used and the color of the medium changes rapidly and becomes cloudy, there is a possibility of yeast contamination.
- This cannot be resolved with bacterial antibiotics such as P/S or Gentamicin.
- An antibiotic for fungi, Antimycotic (=Fungizone, Amphotericin B), must be used.
3. If P/S was used in the culture medium and all stocks are contaminated and must be kept alive,
- Take out a new stock, wash it with PBS, and centrifuge at a lower rpm (500 rpm) than the usual case (1,200 rpm) to remove as many small microorganisms as possible.
- Add P/S + Gentamicin + Antimycotic (=Fungizone, Amphotericin B) to the culture medium and culture.
- Add antibiotics and culture until no more symptoms of microbial contamination are observed after passage.
- Microorganisms are considered to be removed if no more symptoms of microbial contamination are observed when culture is performed with the antibiotics removed.
4. If the problem is not solved despite these efforts, discard even a stable cell line and create a new cell line.
Cell storage method
When the cell line is not in use, it is frozen and stored in liquid nitrogen.
To store the cell line alive, use a freezing medium for cell storage. Generally, 10% DMSO (Dimethyl sulfoxide) is added to culture medium containing 10-20% FBS, or 10% DMSO is added to 90% FBS. “DMSO is called a cryoprotectant” and prevents cell death by preventing the formation of large, sharp ice crystals in the cells. In most cases, DMSO can be used, but in cases where cell differentiation is induced by DMSO, such as HL60, glycerol is used as a substitute. The
speed of cell freezing is also important. If the cell freezing vial is placed in a freezing container and placed in a -70℃ freezer, the temperature drops at a constant rate of about 1℃ per minute, allowing for safer cell freezing. Once the cells are frozen, they are quickly transferred to a liquid nitrogen storage container for storage.
Cryopreservation of cell line (Source: PDA)
“How does DMSO, which is used for cryopreservation of animal cells, protect cells?”
DMSO is a polar solvent, but it mixes well with water, maintains a homogeneous state, and lowers the freezing/melting temperature of water. While pure water forms sharp and large ice crystals, 10% DMSO forms crystals that are about 10 times smaller and amorphous. For this reason, it prevents damage to cells caused by sharp ice crystals.
DMSO has the property of dissolving polar or nonpolar substances, and at high concentrations at room temperature, it can dissolve cell membrane components and create holes, destroying cells. Therefore, when freezing cells, use a storage medium containing DMSO to quickly suspend and freeze the cells, minimizing the time exposed to room temperature.
When thawing cells, thaw them quickly in a 37°C constant temperature water bath, and dilute them at least 10 times with the medium while there is still some ice left, thereby lowering the concentration of DMSO. If the concentration of DMSO at room temperature is 0.5% or higher, it can cause cell membrane damage. Since the DMSO concentration of the cell cryopreservation solution is 10%, and even if it is diluted 10 times, it will still be 1%, the dilution medium should be used in a cold state, and the medium containing DMSO should be removed quickly after centrifugation at 4°C to minimize cell damage.
In addition, when using cryopreserved cells for passage culture, it is recommended not to use them beyond a certain number of passages. The reason is thought to be to minimize cell mutation or physiological activity decline due to over-culture during passage culture and to use high-quality cell lines.
However, there is no such restriction for cryopreserved cell lines. I think you have experienced that cells that have deteriorated due to continuous passage culture or culture beyond the appropriate confluent level will return to normal if they are cryopreserved and then used again.
As a final question, why do cell lines that have been passaged more than the appropriate number of times become good when cryopreserved and then cultured again?
If cells are cultured for a long time, mutations may occur in some parts, but cells with a high local density may undergo cell cycle arrest due to contact inhibition. It is difficult to obtain the best experimental results by using cells in various states with different growth states and physiological activities during the cell subculture process.
You may have experienced that the transfection efficiency is significantly reduced when cells cultured at a high density of ~90% confluent or higher are used during the subculture process. However, you may also know that such cells can be recovered by freezing and then re-culturing.
“What happens during cell cryopreservation?”
It is believed that the secret to cell recovery during cell line cryopreservation lies in DMSO. DMSO, used when cryopreserving cell lines, is known to induce reversible G1 phase cell cycle arrest at a low concentration of 1–2% and synchronize the physiological state of cells. In addition, it is known to restore contact inhibition and prevent cell death caused by high-density culture.
In summary, in order to obtain highly reproducible experimental results in experiments using cell lines, it is necessary to secure a large number of master cell stocks with cells of the initial passage number after distributing the cell line from the cell line bank, and to use working cell stocks made by culturing these cells to keep the number of subcultures to a minimum.
[Summary]
Why is it necessary to record passage numbers for cancer cells that proliferate indefinitely? Since cell lines can undergo mutations during proliferation, passage number monitoring is necessary to maintain cell characteristics and ensure reproducibility of results.
Why is it necessary to culture cells only to ~90% confluent when subculturing? It is necessary to minimize changes in cell physiological activity due to contact inhibition. Why
is it necessary to subculture cancer cell lines that are not primary cells only to a certain number of passages? As the number of subcultures increases, the possibility of mutations increases, and the physiological and activity states deteriorate and diversify due to differences in local cell density, making it difficult to obtain the best results. Why does the condition
of cell lines that have been subcultured more than an appropriate number of times improve when they are cryopreserved and then cultured again? DMSO, used as a cryopreservative, synchronizes the state of cells through reversible cell cycle arrest at a low concentration of 1-2%, recovers from contact inhibition caused by culture, and prevents apoptosis, thereby resetting and recovering the overall physiological activity state of cells.
References
Cell culture Wikipedia (https://en.wikipedia.org/wiki/Cell_culture)
Yao T., Asayama Y. Animal-cell culture media: History, characteristics and current issues. Reproductive Medicine and Biology 2017; 16:99-117.
Cryopreservation of cell lines (https://www.sigmaaldrich.com/technical-documents/protocols/biology/cryopreservation-of-cell-lines.html).
Fiore M., Zanier R., Degrassi F. Reversible G (1) arrest by dimethyl sulfoxide as a new method to synchronize Chinese hamster cells. Mutagenesis 2002;17(5):419-424.
'Lab skills' 카테고리의 다른 글
| microarray (0) | 2024.10.26 |
|---|---|
| luciferase assay (0) | 2024.10.26 |
| western blot (0) | 2024.10.26 |
| Elisa (4) | 2024.10.26 |
| Biological information provided by scRNA-seq analysis (0) | 2024.10.24 |