Bio Python example code
2024. 11. 27. 10:39ㆍComputational biology
import Bio
print(Bio.__doc__)
Collection of modules for dealing with biological data in Python.
The Biopython Project is an international association of developers
of freely available Python tools for computational molecular biology.
http://biopython.org
DNA sequence handling
from Bio import Entrez
from Bio import SeqIO
Entrez.email = "A.N.Other@example.com"
with Entrez.efetch(
db="nucleotide", rettype="fasta", retmode="text", id="1490011893"
) as handle:
seq_record = SeqIO.read(handle, "fasta")
seq_record
SeqRecord(seq=Seq('ATGGAGGAGATGCTGCCCCTCTTTGAGCCCAAGGGCCGGGTCCTCCTGGTGGAC...GAG', SingleLetterAlphabet()), id='MG727867.1', name='MG727867.1', description='MG727867.1 Synthetic construct Taq DNA polymerase gene, partial cds', dbxrefs=[])sequence number count
taq = seq_record.seq
len(taq)
2496
Calculating GC_content
from Bio.SeqUtils import GC
GC(taq)
67.70833333333333
case conversion
taq.upper()
Seq('ATGGAGGAGATGCTGCCCCTCTTTGAGCCCAAGGGCCGGGTCCTCCTGGTGGAC...GAG', SingleLetterAlphabet())
taq.lower()
Seq('atggaggagatgctgcccctctttgagcccaagggccgggtcctcctggtggac...gag', SingleLetterAlphabet())
DNA sequence Translation, Transcription
taq.transcribe()
Seq('AUGGAGGAGAUGCUGCCCCUCUUUGAGCCCAAGGGCCGGGUCCUCCUGGUGGAC...GAG', RNAAlphabet())taq.translate()
Seq('MEEMLPLFEPKGRVLLVDGHHLAYRTFHALKGLTTSRGEPVQAVYGFAKSLLKA...AKE', ExtendedIUPACProtein())
Molecular weight
from Bio.SeqUtils import molecular_weight
molecular_weight(taq)
771589.062500014
molecular_weight(taq.translate())
94387.32270000054
Codon table
from Bio.Data import CodonTable
print(CodonTable.unambiguous_dna_by_name["Standard"])
Table 1 Standard, SGC0
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--
Tm value calculation
from Bio.SeqUtils import MeltingTemp
from Bio.Seq import Seq
primer_DNA = Seq("ATGGAGGAGATGCTGCCCCTCT")
MeltingTemp.Tm_Wallace(primer_DNA)
70.0
Amino Acid Sequence conversion
from Bio.SeqUtils import seq1, seq3
seq3("MEEMLPLFEPKGRVLLVDGHHLAYRTFHALKGLTTSRGEPVQAVYGFAKSLLKA")
'MetGluGluMetLeuProLeuPheGluProLysGlyArgValLeuLeuValAspGlyHisHisLeuAlaTyrArgThrPheHisAlaLeuLysGlyLeuThrThrSerArgGlyGluProValGlnAlaValTyrGlyPheAlaLysSerLeuLeuLysAla'
seq1(
"MetGluGluMetLeuProLeuPheGluProLysGlyArgValLeuLeuValAspGlyHisHisLeuAlaTyrArgThrPheHisAlaLeuLysGlyLeuThrThrSerArgGlyGluProValGlnAlaValTyrGlyPheAlaLysSerLeuLeuLysAla"
)
'MEEMLPLFEPKGRVLLVDGHHLAYRTFHALKGLTTSRGEPVQAVYGFAKSLLKA'
Weblogo
from Bio import AlignIO
from Bio.motifs import Motif
from Bio import motifs
from Bio.Seq import Seq
from Bio.Alphabet import IUPAC
from IPython.display import Image
alignment = AlignIO.read("./data/HBA.aln", "clustal")
instance = []
for record in alignment:
s = Seq(str(record.seq), IUPAC.protein)
instance.append(s)
m = motifs.create(instance)
Motif.weblogo(m, "HBA_logo.png")
Image("HBA_logo.png")

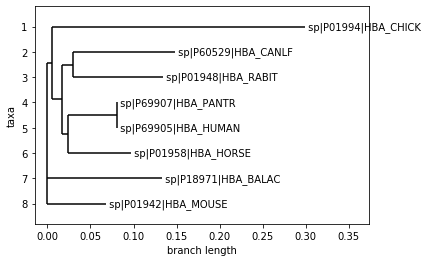
phylogenetic tree
%matplotlib inline
from Bio import Phylo
tree = Phylo.read("./data/HBA.newick", "newick")
Phylo.draw(tree)
Entrez data base search
from Bio import Entrez
Entrez.email = "your@email.com"
handle = Entrez.esearch(db="pubmed", term="machine learning")
record = Entrez.read(handle)
print("Pubmed에 machine learning를 검색하면 총 {}개의 결과".format(record["Count"]))
Pubmed에 machine learning를 검색하면 총 29978개의 결과
KEGG API
from Bio.KEGG import REST
human_pathways = REST.kegg_list("pathway", "hsa").read()
pathways = []
for line in human_pathways.rstrip().split("\n"):
entry, description = line.split("\t")
if "repair" in description.lower():
pathways.append(entry)
print(entry, description)
print(pathways)
path:hsa03410 Base excision repair - Homo sapiens (human)
path:hsa03420 Nucleotide excision repair - Homo sapiens (human)
path:hsa03430 Mismatch repair - Homo sapiens (human)
['path:hsa03410', 'path:hsa03420', 'path:hsa03430']
genes = []
for pathway in pathways:
pathway_file = REST.kegg_get(pathway).read()
current_section = None
for line in pathway_file.rstrip().split("\n"):
section = line[:12].strip()
if not section == "":
current_section = section
if current_section == "GENE":
gene_identifiers, gene_description = line[12:].split("; ")
gene_id, gene_symbol = gene_identifiers.split()
if gene_symbol not in genes:
genes.append(gene_symbol)
print("pathway에 연관된 유전자는 {} 이다".format(",".join(genes)))
pathway에 연관된 유전자는 OGG1,RBX1,SSBP1 이다
from partrita git hub
'Computational biology' 카테고리의 다른 글
| Python-Basic (1) | 2024.11.27 |
|---|---|
| biopython (0) | 2024.11.27 |
| python libraries (1) | 2024.11.27 |
| scRNA-seq Raw Data Preprocessing: scRNA-seq quality control (0) | 2024.11.27 |
| R packages (0) | 2024.11.27 |